


The paraganglion system, a component of the neuroendocrine system, comprises the adrenal medullae and the extra-adrenal paraganglion system. Tissues of the extra-adrenal system have been classified into 4 groups: branchiomeric (associated with arterial vessels and cranial nerves derived from the branchial arches); intravagal; aorticosympathetic; and visceral-autonomic (including cardiac atria, bladder, liver hilum and mesenteries).1 Tumours, or paragangliomas, may arise anywhere in this system. Paragangliomas have also been reported to have arisen in locations where paraganglia are not normally found, such as the duodenum,2 stomach,3 vulva,4 spinal cord,5 filum terminale6 and cauda equina.6 The majority of paragangliomas are nonfunctional and benign; however, they may secrete catecholamines or have malignant potential.1
Case Report
A 52-year-old man complained of increasing low-back pain over 2 years. The pain began as an odd sensation described as a “deep itch” in the perianal area, accompanied by occasional sacral pain, which radiated down both legs to the knees. This tingling pain became constant, was increasing in intensity and had recently been interfering with his sleep. There was no history of low-back pain, no precipitating trauma and no aggravating or alleviating factors.
The patient appeared healthy and had a blood pressure of 130/70 mm Hg. Examination of the spine showed a normal range of motion. There was tenderness to palpation over the sacral region. Lower extremity examination indicated normal motor and sensory function. No changes in bowel, bladder or sexual function were reported.
Radiographs of the lumbar spine and sacrum showed a radiolucent area in the body of the sacrum. Bone scanning showed a large area of increased uptake in the sacrum with a central area of decreased uptake. Computed tomography (CT) indicated a large, expanding, destructive mass involving the mid-sacrum (Fig. 1). CT-guided needle core biopsy gave a histologic diagnosis of paraganglioma.

Transverse (axial) computed tomography (CT) at S2 to S3 showing an expansile mass within the body of the sacrum.
Magnetic resonance imaging (MRI) demonstrated a 5 × 5 × 6-cm soft-tissue mass centred in the body of the sacrum at S3 (Fig. 2). The mass extended anteriorly and posteriorly through both cortices of the sacrum to involve the presacral fat anteriorly and the thoracolumbar fascia posteriorly. The mass was lobulated and well defined, and there was no evidence of invasion of pelvic structures, subcutaneous fat or skin. CT of the chest, abdomen and pelvis showed no evidence of other masses, consistent with a primary intraosseous sacral tumour. Results of routine laboratory investigations were all within normal limits.
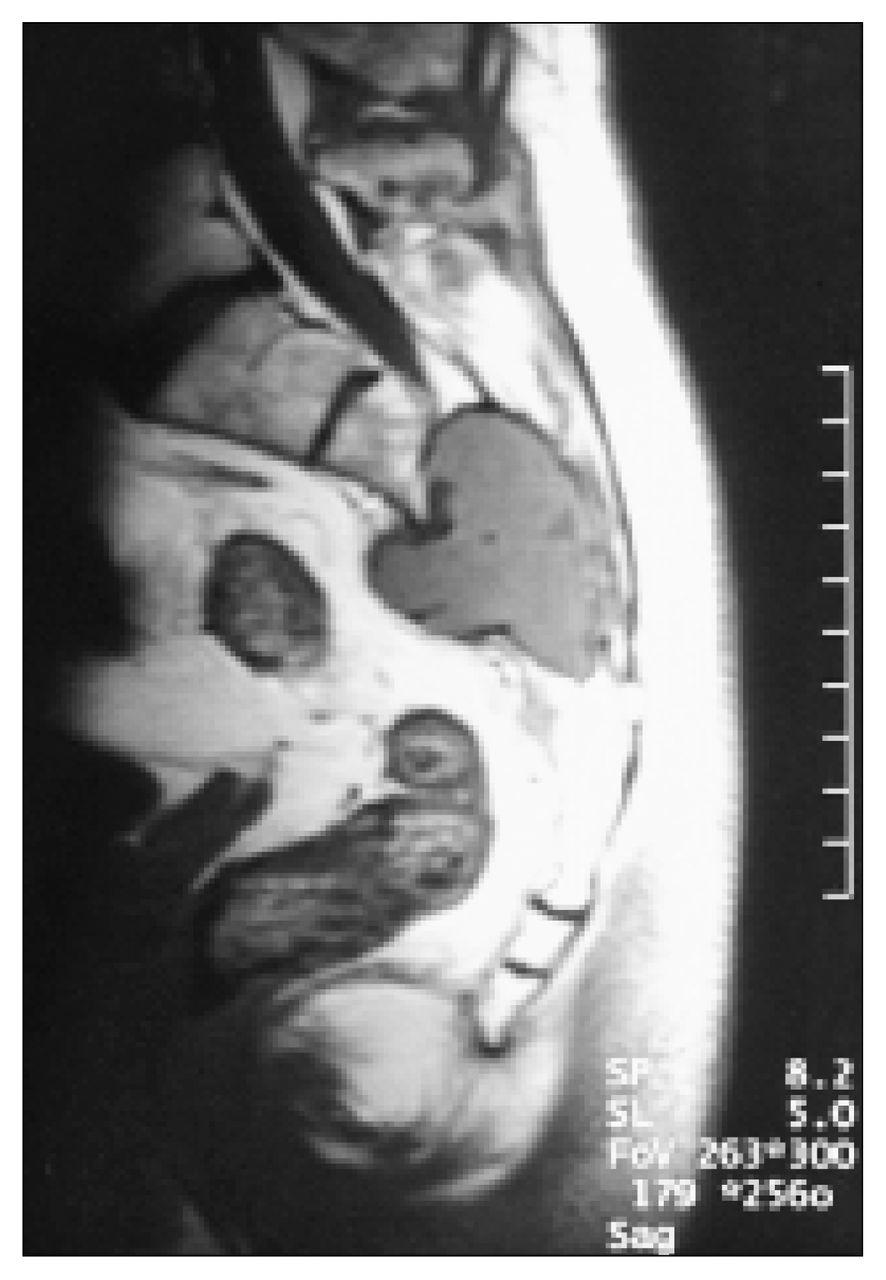
Sagittal magnetic resonance T1-weighted image revealing the sacral mass eroding through the anterior and posterior cortices of the sacral body.
Based on the histologic diagnosis, we decided to excise the tumour. Surgical resection was attempted through a posterior midline incision, exposing a vascular, reddish-brown, pulsatile mass protruding posteriorly from a 2 × 3-cm defect in the sacrum. Because of the potential for significant blood loss, this attempt was aborted. A small specimen of the tumour revealed the same histologic pattern as the previous needle core biopsy specimen.
The patient was scheduled for further resection 6 weeks later. At angiography, performed 24 hours preoperatively, the middle sacral artery and right and left lateral sacral arteries were embolized by injection of large polyvinyl alcohol particles, rendering the tumour almost entirely avascular. At operation, the tumour appeared shrunken and no longer pulsatile. The defect in the posterior cortex of the sacral body was enlarged to expose the entire tumour. The S2 and S3 nerve roots were identified and spared. The tumour lifted off the anterior cortex of the sacrum relatively easily, revealing the defect in the anterior cortex where the tumour had extended anteriorly. The mass was removed. Small pockets of tumour remaining in the interface between tumour and sacrum were curetted and cauterized. The procedure was well tolerated, and there was no need for blood transfusion. Postoperative recovery was uncomplicated, and the patient was discharged home 5 days postoperatively.
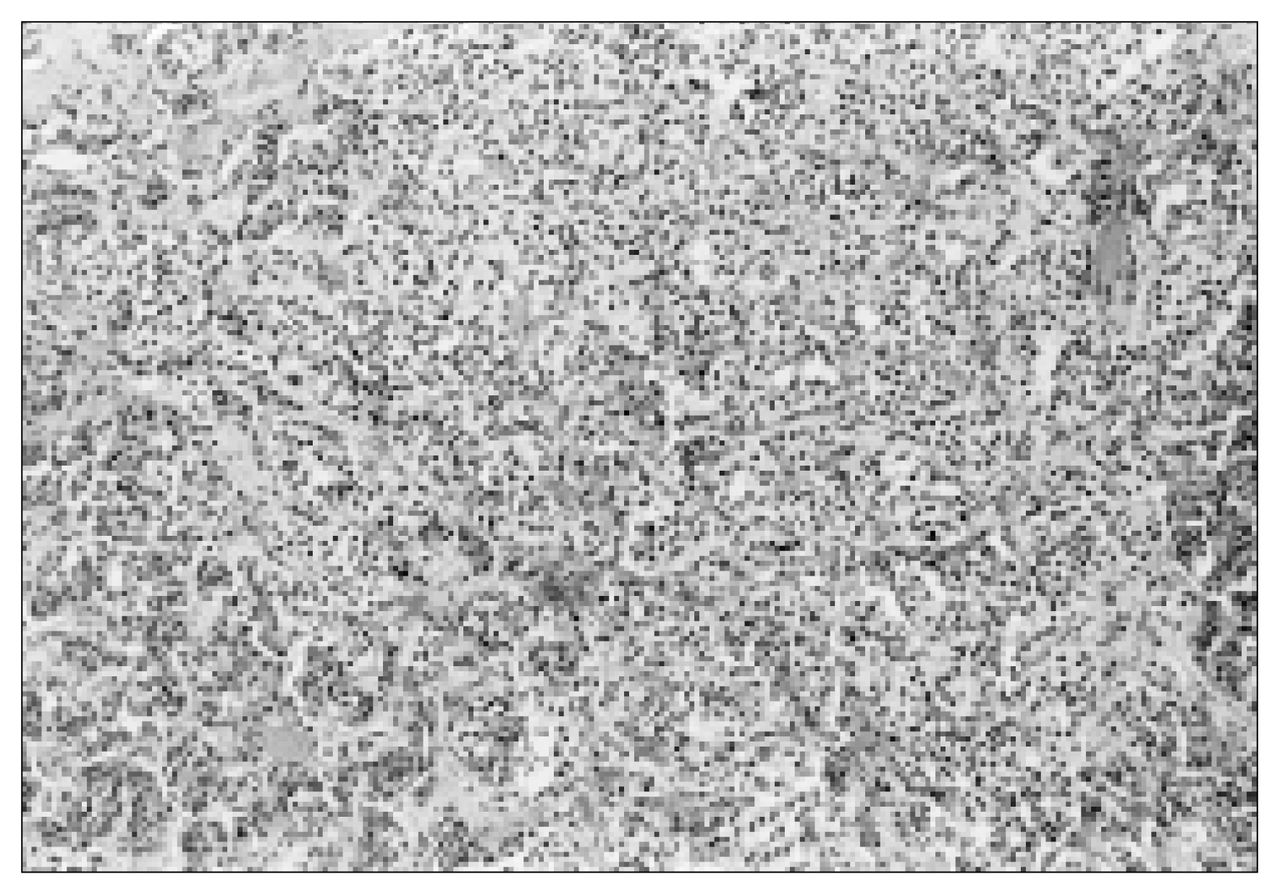
The histologic characteristics of this tumour were typical of paragangliomas. The tumour was composed of tightly clustered nests of cells divided by delicate connective tissue septae (“Zellballen” pattern) and possessed a rich vascular network (Fig. 3).1 Tumour cells were large, epithelioid and contained round nuclei demonstrating some variation in size (Fig. 4). On immunohistochemical analysis, tumour cells were found to be positive for vimentin, neuron specific enolase and chromogranin. Scattered S-100 positive cells were identified.
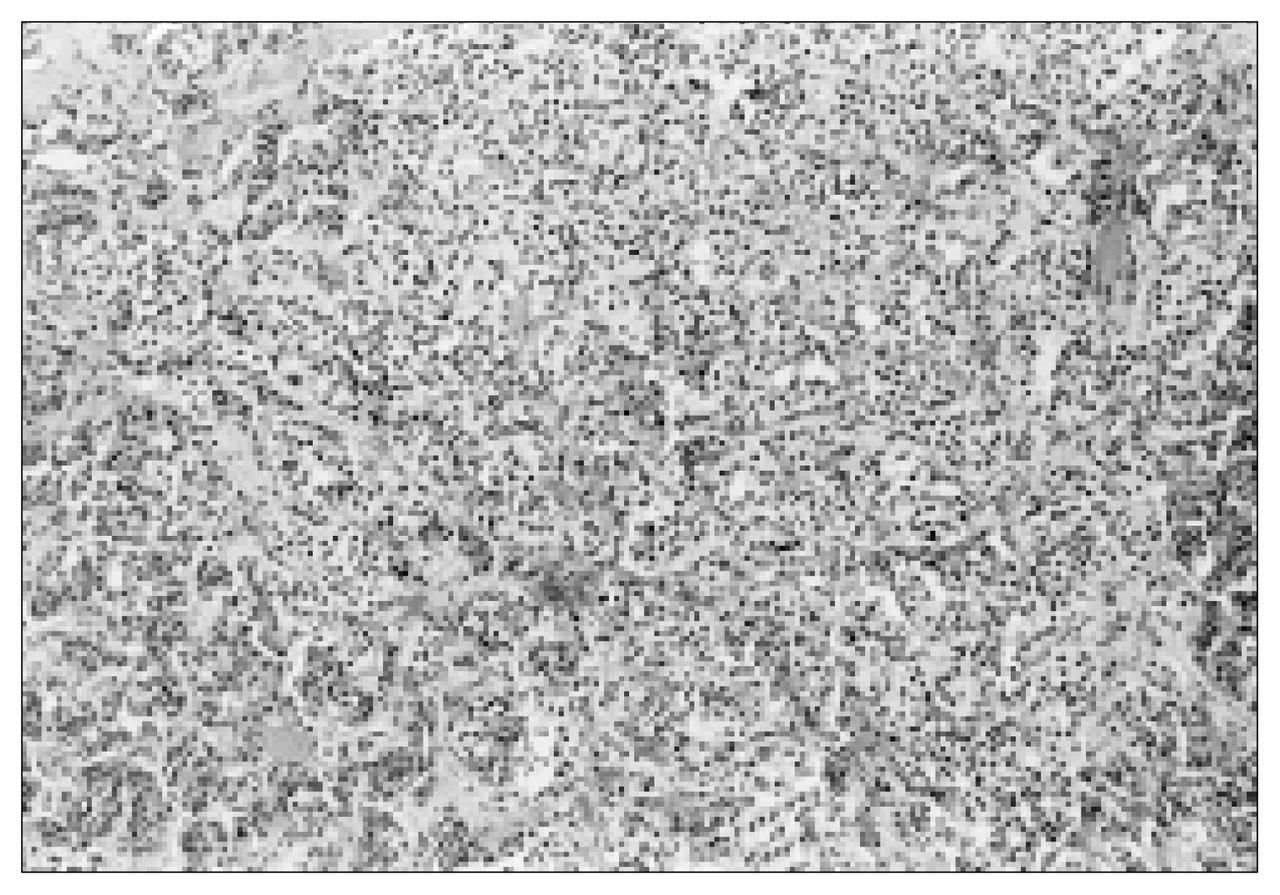
Tightly clustered nests of tumour cells divided by delicate connective tissue septae (“Zellballen” pattern) (hematoxylin–eosin stain; original magnification × 40).

Large, epithelioid tumour cells containing round nuclei demonstrating some variation in size (hematoxylin–eosin stain; original magnification × 160).
Follow-up at 2 years showed complete resolution of preoperative symptoms, and magnetic resonance imaging revealed no evidence of local recurrence. In addition, Indium-111-octreotide scintigraphy7 performed 1 year after resection, also showed no evidence of local recurrence and no other primary or secondary paragangliomas.
Discussion
Paragangliomas are neoplasms derived from neurosecretory cells believed to be of neural crest origin.1 These lesions are relatively uncommon and tend to occur in a variety of locations normally rich in paraganglionic tissue, including arteries, vagal system, sympathetic chain and certain viscera. 1 The cellular architecture of these neoplasms is similar to that of normal paraganglionic tissues, with the predominant cell being the type I, or chief cell, arranged in compact nests (“Zellballen” pattern).1 Interspersed type II, or sustentacular cells, which are S-100 positive8 are also present within these neoplasms.1
A MEDLINE search of articles published between 1966 and 1997 (key words “paraganglioma” or “neuroendocrine tumour”) was conducted. Paragangliomas associated with the spine are unusual, with fewer than 150 cases previously reported in the literature. Most of these have been intradural, extramedullary lesions in the lumbar region. Cases have been reported as cephalad as T5, with others extending as far caudad as S1.9 Only 2 studies have reported extradural extension from an intradural lesion.6,10 Vertebral erosion as a result of compression by adjacent tumour has been reported;6 however, to our knowledge, no case of a paraganglioma originating primarily within bone has been published.
This is the first report of an intraosseous paraganglioma, centred within the sacral body. The anterior and posterior cortices of the sacral body were eroded with extraosseous extension of the tumour. This lesion was clearly extradural, and although it encroached upon the S2 and S3 nerve roots posteriorly, it did not appear to arise from the sacral nerve roots or the filum terminale. The histologic features were characteristic of a paraganglioma. CT of the chest and abdomen, as well as 111Inoctreotide scintigraphy showed no evidence of metastases or other primary lesion. Two years postoperatively, there was no evidence of local recurrence on MRI.
The reported occurrence of paragangliomas outside the usual distribution of paraganglionic tissue may be a reflection of the migratory and differentiation properties of neural crest cells during embryogenesis. Abnormalities in these processes may produce ectopic nests of cells capable of neoplastic transformation.
The differential diagnosis of a lytic lesion of the sacrum must include chordoma,11 aneurysmal bone cyst,12 and giant cell tumour,13 as well as osteoblastoma,14 chondrosarcoma,15 hemangiopericytoma,16 lymphoma,17 multiple myeloma,17 or metastatic disease. Tumours such as carcinoids,8 plasmacytoma,18 chondroblastoma,19 mesenchymal chondrosarcoma,20 myxopapillary ependymoma,21 and giant sacral schwannoma, 22 have rarely been reported in the sacrum and should also be considered. Although rare, paraganglioma should now be considered in the differential diagnosis of a destructive lesion of the sacrum.
- Accepted October 7, 1998.
References
In this issue
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.